




Abstract
Coronary heart disease (CHD) is a leading cause of death worldwide, yet many areas of its pathogenesis remain unknown or poorly understood, leaving potential for novel preventive and therapeutic interventions. Recent major advances in genomic science and technology have opened new avenues of investigation in the pathogenesis of CHD, some of which are leading to clinical translation.
The published literature in CHD genetics has burgeoned in the last 5 years with the reporting of genome-wide association studies (GWASs) and many other findings.
Identification of many genetic variants with small effects on CHD risk has been a common finding. These have included several predicted loci, such as those involved in conventional CHD risk factors (e.g. plasma lipids) and many novel loci, where their mechanism of action is unclear. The need for large, collaborative approaches to research has also become clear and is now an accepted modus operandi.
The clinical utility of novel GWAS findings remains uncertain. In particular, the relative contribution of common variants of modest effect and rare variants of larger effects to risk of CHD or response to drugs is unclear.
As a greater number of larger GWASs are conducted in CHD and its related phenotypes, much effort is being made to find translational applications for their findings. Therapeutics, prediction and pathology are major areas of research endeavour.
Introduction: the pathology of and the genetic architecture of coronary heart disease
Coronary heart disease (CHD) has both genetic and environmental precipitants and is a leading cause of morbidity and mortality globally.1 The major risk factors for the development of CHD are male gender, increasing age, elevated levels of plasma low-density lipoprotein cholesterol (LDL-C), elevated blood pressure, obesity, with life style factors such as smoking, a high fat diet and lack of exercise, all making a contribution. High plasma LDL-C concentration drives development of coronary atherosclerotic plaques, which can rupture, leading to vessel occlusion and ischaemic events. The lifetime risk of CHD is around 50% in men and 33% in women, with around 96% of all events occurring after 50 years of age.2 The burden of atherosclerosis increases with age and its long preclinical phase provides a window of opportunity for risk prediction and CHD prevention.
The genetic architecture of CHD has been widely studied, and understanding of the genetic basis of the pathogenesis of CHD has advanced substantially in the last decade. In part, this is a result of major advances in genotyping technology and genome variation databases developed on the basis of the Human Genome project. It is also because many established longitudinal cohorts and case–control studies worldwide are undertaking genetic investigations within their research programmes, and the data, much of which are publically available, are being analysed in large consortia leading to many new discoveries. There are, however, still large gaps in our understanding of how CHD develops, and, importantly, how causal pathways might be exploited as targets for prevention through pharmacological, behavioural or policy interventions. Here, we discuss the recent advances in investigating the genetics of CHD, and their paths to clinical translation.
Lessons from monogenic coronary disease
When considering the genetic basis of CHD, a distinction is necessary between its rarer, monogenic causes, and the common (also termed ‘polygenic’) forms whose pathogenesis involves several factors—genetics, behaviour and the environment. While polygenic CHD is of greatest interest since it places a large burden on the public health, much of the foundation of our understanding of the role of common genetic variation has arisen from the investigation of monogenic disease, characterized by low-frequency genetic variants with large phenotypic effects. Indeed, research into monogenic CHD preceded work on polygenic coronary disease by a number of decades.
An important and illustrative example of a monogenic disease predisposing to early CHD is familial hypercholesterolaemia (FH). FH is characterized by very high plasma concentrations of LDL-C, early incidence of myocardial infarction (MI) and cutaneous lipid deposition (xanthomata and xanthelasmata).3 FH follows an autosomal dominant pattern of inheritance, and is known to be caused by mutations in the genes encoding the LDL receptor (LDLR),4 the major protein of the LDL-C particle and the ligand for the receptor, apolipoprotein B (APOB),5,6 or proprotein convertase subtilisin/kexin type 9 (PCSK9), a protein involved in the degradation of LDLRs.7 Individuals with FH in the age range of 20–59 years have a standardized mortality ratio of ∼8 compared with the general population,8 mainly due to CHD, which can be reduced to the population level of risk by early and intensive statin treatment.9 Although FH is thought to occur in ∼1 in 500 of the population in most countries, it probably accounts for only around 5% of early CHD events (i.e. those occurring before the age of 45).10 Routine clinical genetic diagnostic methods have been established11 that can detect the causative mutation in up to 80% of patients in whom clinical suspicion of the disorder is greatest, and DNA testing in patients and their families using mutation information has been recommended in the UK by the National Institute for Health and Clinical Excellence (NICE)12 and has been actively implemented in several other countries.13–15 Once the mutation has been identified, relatives of the patient can be tested, and, if found to be carriers, can be offered lifestyle advice and early treatment with statins to reduce their very high risk of future CHD.
The ‘revolutions’ of the post-genome era
Early investigations into the genetic variants that predispose to development of CHD, such as those in FH, relied on candidate gene methods, whereby frequencies of a small number of selected genetic variants were compared between groups of cases and controls. The small samples used in such studies resulted in a number of false-positive findings, and led to substantial publication bias. Furthermore, many early reported associations have not been replicated. However, as shown in Table 1, these studies did identify several variants associated with CHD risk that have been subsequently replicated and validated.16 These data confirm that common genetic variation in genes involved in lipid metabolism is likely to be a causal factor in CHD.
Previously identified lipid-associated candidate gene loci confirmed by GWA studies for their association with the risk of CHD.
| Locus | Gene | Lipid trait |
|---|---|---|
| 1p32.3 | PCSK9 | LDL-C |
| 1p31.3 | ANGPTL3 | Triglycerides |
| 2p24.1 | APOB | LDL-C |
| 5q13.3 | HMGCR | LDL-C |
| 6q25.3 | LPA | Lipoprotein(a) |
| 8p21.3 | LPL | Triglycerides |
| 9q31.1 | ABCA1 | HDL-C |
| 11q12.2 | FADS1/FADS2/FADS3 | Triglycerides |
| 11q23.3 | APOA1 | Triglycerides |
| 12q24.31 | HNF1A | Total cholesterol |
| 15q21.3 | LIPC | HDL-C |
| 16q13 | CETP | HDL-C |
| 16q22.1 | LCAT | HDL-C |
| 18q21.1 | LIPG | HDL-C |
| 19p31,2 | ANGPTL-4 | HDL-C |
| 19p23.3 | LDLR | LDL-C |
| 19q13.32 | APOE | LDL-C |
| 20q13.12 | PLTP | HDL-C |
| Locus | Gene | Lipid trait |
|---|---|---|
| 1p32.3 | PCSK9 | LDL-C |
| 1p31.3 | ANGPTL3 | Triglycerides |
| 2p24.1 | APOB | LDL-C |
| 5q13.3 | HMGCR | LDL-C |
| 6q25.3 | LPA | Lipoprotein(a) |
| 8p21.3 | LPL | Triglycerides |
| 9q31.1 | ABCA1 | HDL-C |
| 11q12.2 | FADS1/FADS2/FADS3 | Triglycerides |
| 11q23.3 | APOA1 | Triglycerides |
| 12q24.31 | HNF1A | Total cholesterol |
| 15q21.3 | LIPC | HDL-C |
| 16q13 | CETP | HDL-C |
| 16q22.1 | LCAT | HDL-C |
| 18q21.1 | LIPG | HDL-C |
| 19p31,2 | ANGPTL-4 | HDL-C |
| 19p23.3 | LDLR | LDL-C |
| 19q13.32 | APOE | LDL-C |
| 20q13.12 | PLTP | HDL-C |
Previously identified lipid-associated candidate gene loci confirmed by GWA studies for their association with the risk of CHD.
| Locus | Gene | Lipid trait |
|---|---|---|
| 1p32.3 | PCSK9 | LDL-C |
| 1p31.3 | ANGPTL3 | Triglycerides |
| 2p24.1 | APOB | LDL-C |
| 5q13.3 | HMGCR | LDL-C |
| 6q25.3 | LPA | Lipoprotein(a) |
| 8p21.3 | LPL | Triglycerides |
| 9q31.1 | ABCA1 | HDL-C |
| 11q12.2 | FADS1/FADS2/FADS3 | Triglycerides |
| 11q23.3 | APOA1 | Triglycerides |
| 12q24.31 | HNF1A | Total cholesterol |
| 15q21.3 | LIPC | HDL-C |
| 16q13 | CETP | HDL-C |
| 16q22.1 | LCAT | HDL-C |
| 18q21.1 | LIPG | HDL-C |
| 19p31,2 | ANGPTL-4 | HDL-C |
| 19p23.3 | LDLR | LDL-C |
| 19q13.32 | APOE | LDL-C |
| 20q13.12 | PLTP | HDL-C |
| Locus | Gene | Lipid trait |
|---|---|---|
| 1p32.3 | PCSK9 | LDL-C |
| 1p31.3 | ANGPTL3 | Triglycerides |
| 2p24.1 | APOB | LDL-C |
| 5q13.3 | HMGCR | LDL-C |
| 6q25.3 | LPA | Lipoprotein(a) |
| 8p21.3 | LPL | Triglycerides |
| 9q31.1 | ABCA1 | HDL-C |
| 11q12.2 | FADS1/FADS2/FADS3 | Triglycerides |
| 11q23.3 | APOA1 | Triglycerides |
| 12q24.31 | HNF1A | Total cholesterol |
| 15q21.3 | LIPC | HDL-C |
| 16q13 | CETP | HDL-C |
| 16q22.1 | LCAT | HDL-C |
| 18q21.1 | LIPG | HDL-C |
| 19p31,2 | ANGPTL-4 | HDL-C |
| 19p23.3 | LDLR | LDL-C |
| 19q13.32 | APOE | LDL-C |
| 20q13.12 | PLTP | HDL-C |
The sequencing of the human genome17,18 and subsequent mapping of single nucleotide polymorphisms (SNPs) by the International HapMap Consortium19 represented a revolution for investigating the genetics of common, complex diseases such as CHD and provided a platform for the development of genome-wide association studies (GWASs). In contrast to candidate gene studies that aim to test specific hypotheses based on pre-specified genes, GWASs are hypothesis free, and simultaneously test associations of large numbers of variants across the genome with phenotypes that may be dichotomous (such as the presence of CHD), or continuous such as blood pressure, circulating lipid concentrations or carotid artery intima-media thickness (Fig. 1a and b). Typically, samples of several thousand individuals are genotyped using chip-based platforms that simultaneously type a large number of common genetic variants (normally 500 000–1 million) in a quick and efficient process. Since measuring each genetic marker in each study participant represents an individual statistical test, the likelihood of false-positive associations is high, and stringent criteria for declaration of true-positive associations are needed. These criteria typically take the form of P-value thresholds of P < 5 × 10−8, which is termed ‘genome-wide significance’. The modest phenotypic effects of common genetics variants20 coupled with the stringent statistical criteria, necessitate very large sample sizes to detect true-positive effects, which has led to the formation of large, international research consortia, bringing together existing and newly established studies to maximize statistical power. All SNPs showing a P-value below the pre-specified significance threshold for their association with the phenotype in the initial discovery phase of a GWAS are then further examined in a ‘replication’ phase in one or more independent samples, to overcome bias such as the winner's curse21 and avoid false-positive associations. As such, the variants discovered with this methodology are perhaps some of the most statistically robust findings in the biomedical literature.
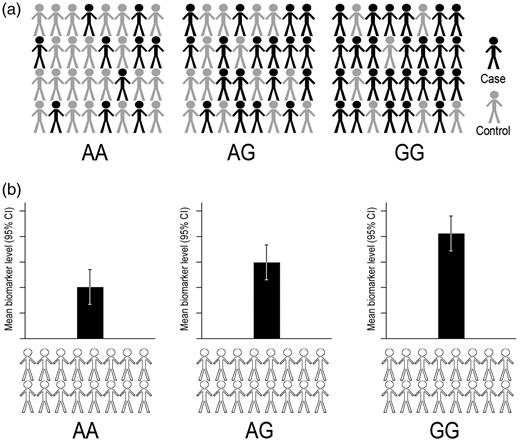
A GWAS design. (a) A case control design for identification of variants associated with risk of a binary endpoint (e.g. CHD). Evaluating differences in proportions of cases and controls between genotype classes for a given SNP (i.e. AA, AG and GG) allow the estimation of association of that SNP with the risk of disease. (b) Identification of variants associated with a continuous biomarker (e.g. plasma LDL-C) by comparison of biomarker levels according to genotype class.
However, although over 10 million SNPs have been discovered through large-scale resequencing efforts such as the 1000 Genomes project, not all of these SNPs need to be directly genotyped to achieve genome-wide coverage. Knowledge of linkage disequilibrium (LD, the non-random association of two or more loci with one another) throughout the genome allows ‘imputation’ of un-typed variants from large reference panels which can be performed with a high degree of statistical certainty. Whilst such imputation has been used to successfully fine-map causal variants in loci identified by GWAS,22 this technique has also proved enormously useful in meta-analyses of GWAS that have used different genotyping platforms. Using such an approach to analyse risk of sick sinus syndrome led to identification of an un-typed likely causative SNP in a previously unidentified susceptibility gene, MYH6, encoding the alpha heavy chain subunit of cardiac myosin.23
Despite the strengths of the GWAS approach, it does have limitations. First, the risk variants identified by GWA studies have, in general, modest phenotypic effects. For example, odds ratios between 1.05 and 1.30 for CHD are typically associated with the carriage of each copy of the risk allele of a given SNP, when compared with the carriage of none. When considered alone, most SNPs therefore explain only a very small proportion of the population variance in a phenotype, which is in keeping with the polygenic model of pathogenesis.24 To overcome this limitation, investigators are beginning to use ‘gene scores’ that combine data on a number of phenotype-associated genetic variants in order to explain a larger proportion of variance, and so increase the statistical power. Such scores have been used in relation to genetic determinants of structural cardiac phenotypes,25 plasma lipid concentration26 and risk of type II diabetes27 and CHD.28 For example, a gene score of 20 LDL-C raising SNPs has been reported to explain 14% of LDL-C variance in healthy men and women.26 Men carrying 22 or fewer LDL-C raising alleles (the 5% of the population with lowest LDL-C levels) having a mean LDL-C of 3.5 mmol/l, while those with 33 or more raising alleles (the 5% of the population with highest LDL-C levels) have a mean LDL-C of 5.0 mmol/l. The potential clinical utility of such gene scores or their use to explore causality is discussed below.
Second, because genotyping platforms do not detect every SNP in the genome, the variants reported by a GWAS are often not the true causal variant at the cognate locus, but, through LD, act as proxies for one or more causal variant(s). As a consequence, identification of the causal variants and elucidation of their true biological function has proved a major challenge, and research in this area of translation is an important hurdle in the exploitation of GWAS findings. Finally, GWAS genotyping platforms generally include only SNPs that occur relatively commonly in the population, typically with minor allele frequency (MAF) >5%. GWASs are therefore poorly equipped to detect low-frequency variants (MAF <5%) that may have larger phenotypic effects, though they may be imputable as discussed above. Although rare SNPs may be of clinical importance for the individual carrying them and their relatives (for example, the variants causing FH), these variants, being rare, are unlikely to explain a large proportion of phenotypic variance on the level of the whole population.
GWA studies and coronary heart disease
In 2007 the field of cardiovascular genetics was ignited by the simultaneous publication of four separate GWASs of CHD risk.29–32 Each of these reported, among other loci, a strong association between common variants on chromosome 9p21 and risk of CHD, and many other investigators have replicated the strong association with CHD.33 The identified SNPs are commonly found in the population (MAF = 40–50%) and each copy of the risk allele for the identified SNP conferred 30–40% higher risk of CHD. These data highlighted a strength of the GWAS method, as the variants are located in a gene desert—an area of the genome unlikely to be addressed by candidate gene studies. Interestingly, carriage of the risk allele was not associated with conventional cardiovascular risk factors such as lipids, blood pressure or obesity, suggesting a novel mechanism for the effect of the variant.
Following these initial publications, a further 12 GWASs of CHD have been reported (www.genome.gov/gwastudies, accessed January 2012). As shown in Table 2, a recently published meta-analysis of 14 CHD GWASs, including 22 233 cases and 56 682 controls validated the association of 10 previously reported loci, and identified 13 new variants robustly associated with CHD.34
Novel loci identified by the CARDIoGRAM GWAS for their association with risk of CHD.
| Locus | Gene(s) | Odds ratio (95% CI) | Cardiovascular risk factor associations | Associations in GWASs of other traits |
|---|---|---|---|---|
| 9p21.3 | CDKN2A, CDKN2B | 1.29 (1.23–1.36) | — | AAA, intracranial aneurysm, type II diabetes |
| 21q22.11 | MRPS6 | 1.18 (1.12–1.24) | — | Respiratory function |
| 1p32.2 | MIA3 | 1.14 (1.09–1.20) | — | — |
| 2q33.1 | WDR12 | 1.14 (1.09–1.19) | — | — |
| 11q23.3 | ZNF259, APOA5-A4-C3-A1 | 1.13 (1.19–1.16) | Trigs, HDL-C | Metabolic syndrome |
| 10q21.32 | CYP17A1, CNNM2, NT5C2 | 1.12 (1.08–1.16) | Hypertension | — |
| 3q22.3 | MRAS | 1.12 (1.07–1.16) | — | — |
| 1q13.3 | SORT1 | 1.11 (1.08–1.15) | LDL-C | Chronic kidney disease |
| 9q34.2 | ABO | 1.10 (1.07–1.13) | — | ICAM-1, E-selectin |
| 6p24.1 | PHACTR1 | 1.10 (1.06–1.13) | — | — |
| 10q11.21 | CXCL12 | 1.09 (1.07–1.13) | — | — |
| 7q32.2 | ZC3HC1 | 1.09 (1.07–1.12) | — | — |
| 6q23.2 | TCF21 | 1.08 (1.06–1.10) | — | — |
| 15q25.1 | ADAMTS7 | 1.08 (1.06–1.10) | — | — |
| 6p21.32 | ANKS1A | 1.07 (1.05–1.10) | — | — |
| 14q32.2 | HHIPL1 | 1.07 (1.05–1.10) | — | — |
| 13q34 | COL4A1, COL4A2 | 1.07 (1.05–1.09) | — | Psychiatric disorders |
| 17p13.3 | SMG6, SRR | 1.07 (1.05–1.09) | — | Ascending aortic diameter |
| 17p11.2 | RASD1, SMCR3, PEMT | 1.07 (1.05–1.09) | — | Parkinson's disease |
| 12q24.12 | SH2B3 | 1.07 (1.04–1.10) | Platelet/WBC | Type I diabetes |
| Locus | Gene(s) | Odds ratio (95% CI) | Cardiovascular risk factor associations | Associations in GWASs of other traits |
|---|---|---|---|---|
| 9p21.3 | CDKN2A, CDKN2B | 1.29 (1.23–1.36) | — | AAA, intracranial aneurysm, type II diabetes |
| 21q22.11 | MRPS6 | 1.18 (1.12–1.24) | — | Respiratory function |
| 1p32.2 | MIA3 | 1.14 (1.09–1.20) | — | — |
| 2q33.1 | WDR12 | 1.14 (1.09–1.19) | — | — |
| 11q23.3 | ZNF259, APOA5-A4-C3-A1 | 1.13 (1.19–1.16) | Trigs, HDL-C | Metabolic syndrome |
| 10q21.32 | CYP17A1, CNNM2, NT5C2 | 1.12 (1.08–1.16) | Hypertension | — |
| 3q22.3 | MRAS | 1.12 (1.07–1.16) | — | — |
| 1q13.3 | SORT1 | 1.11 (1.08–1.15) | LDL-C | Chronic kidney disease |
| 9q34.2 | ABO | 1.10 (1.07–1.13) | — | ICAM-1, E-selectin |
| 6p24.1 | PHACTR1 | 1.10 (1.06–1.13) | — | — |
| 10q11.21 | CXCL12 | 1.09 (1.07–1.13) | — | — |
| 7q32.2 | ZC3HC1 | 1.09 (1.07–1.12) | — | — |
| 6q23.2 | TCF21 | 1.08 (1.06–1.10) | — | — |
| 15q25.1 | ADAMTS7 | 1.08 (1.06–1.10) | — | — |
| 6p21.32 | ANKS1A | 1.07 (1.05–1.10) | — | — |
| 14q32.2 | HHIPL1 | 1.07 (1.05–1.10) | — | — |
| 13q34 | COL4A1, COL4A2 | 1.07 (1.05–1.09) | — | Psychiatric disorders |
| 17p13.3 | SMG6, SRR | 1.07 (1.05–1.09) | — | Ascending aortic diameter |
| 17p11.2 | RASD1, SMCR3, PEMT | 1.07 (1.05–1.09) | — | Parkinson's disease |
| 12q24.12 | SH2B3 | 1.07 (1.04–1.10) | Platelet/WBC | Type I diabetes |
Data from Schunkert et al.34
Novel loci identified by the CARDIoGRAM GWAS for their association with risk of CHD.
| Locus | Gene(s) | Odds ratio (95% CI) | Cardiovascular risk factor associations | Associations in GWASs of other traits |
|---|---|---|---|---|
| 9p21.3 | CDKN2A, CDKN2B | 1.29 (1.23–1.36) | — | AAA, intracranial aneurysm, type II diabetes |
| 21q22.11 | MRPS6 | 1.18 (1.12–1.24) | — | Respiratory function |
| 1p32.2 | MIA3 | 1.14 (1.09–1.20) | — | — |
| 2q33.1 | WDR12 | 1.14 (1.09–1.19) | — | — |
| 11q23.3 | ZNF259, APOA5-A4-C3-A1 | 1.13 (1.19–1.16) | Trigs, HDL-C | Metabolic syndrome |
| 10q21.32 | CYP17A1, CNNM2, NT5C2 | 1.12 (1.08–1.16) | Hypertension | — |
| 3q22.3 | MRAS | 1.12 (1.07–1.16) | — | — |
| 1q13.3 | SORT1 | 1.11 (1.08–1.15) | LDL-C | Chronic kidney disease |
| 9q34.2 | ABO | 1.10 (1.07–1.13) | — | ICAM-1, E-selectin |
| 6p24.1 | PHACTR1 | 1.10 (1.06–1.13) | — | — |
| 10q11.21 | CXCL12 | 1.09 (1.07–1.13) | — | — |
| 7q32.2 | ZC3HC1 | 1.09 (1.07–1.12) | — | — |
| 6q23.2 | TCF21 | 1.08 (1.06–1.10) | — | — |
| 15q25.1 | ADAMTS7 | 1.08 (1.06–1.10) | — | — |
| 6p21.32 | ANKS1A | 1.07 (1.05–1.10) | — | — |
| 14q32.2 | HHIPL1 | 1.07 (1.05–1.10) | — | — |
| 13q34 | COL4A1, COL4A2 | 1.07 (1.05–1.09) | — | Psychiatric disorders |
| 17p13.3 | SMG6, SRR | 1.07 (1.05–1.09) | — | Ascending aortic diameter |
| 17p11.2 | RASD1, SMCR3, PEMT | 1.07 (1.05–1.09) | — | Parkinson's disease |
| 12q24.12 | SH2B3 | 1.07 (1.04–1.10) | Platelet/WBC | Type I diabetes |
| Locus | Gene(s) | Odds ratio (95% CI) | Cardiovascular risk factor associations | Associations in GWASs of other traits |
|---|---|---|---|---|
| 9p21.3 | CDKN2A, CDKN2B | 1.29 (1.23–1.36) | — | AAA, intracranial aneurysm, type II diabetes |
| 21q22.11 | MRPS6 | 1.18 (1.12–1.24) | — | Respiratory function |
| 1p32.2 | MIA3 | 1.14 (1.09–1.20) | — | — |
| 2q33.1 | WDR12 | 1.14 (1.09–1.19) | — | — |
| 11q23.3 | ZNF259, APOA5-A4-C3-A1 | 1.13 (1.19–1.16) | Trigs, HDL-C | Metabolic syndrome |
| 10q21.32 | CYP17A1, CNNM2, NT5C2 | 1.12 (1.08–1.16) | Hypertension | — |
| 3q22.3 | MRAS | 1.12 (1.07–1.16) | — | — |
| 1q13.3 | SORT1 | 1.11 (1.08–1.15) | LDL-C | Chronic kidney disease |
| 9q34.2 | ABO | 1.10 (1.07–1.13) | — | ICAM-1, E-selectin |
| 6p24.1 | PHACTR1 | 1.10 (1.06–1.13) | — | — |
| 10q11.21 | CXCL12 | 1.09 (1.07–1.13) | — | — |
| 7q32.2 | ZC3HC1 | 1.09 (1.07–1.12) | — | — |
| 6q23.2 | TCF21 | 1.08 (1.06–1.10) | — | — |
| 15q25.1 | ADAMTS7 | 1.08 (1.06–1.10) | — | — |
| 6p21.32 | ANKS1A | 1.07 (1.05–1.10) | — | — |
| 14q32.2 | HHIPL1 | 1.07 (1.05–1.10) | — | — |
| 13q34 | COL4A1, COL4A2 | 1.07 (1.05–1.09) | — | Psychiatric disorders |
| 17p13.3 | SMG6, SRR | 1.07 (1.05–1.09) | — | Ascending aortic diameter |
| 17p11.2 | RASD1, SMCR3, PEMT | 1.07 (1.05–1.09) | — | Parkinson's disease |
| 12q24.12 | SH2B3 | 1.07 (1.04–1.10) | Platelet/WBC | Type I diabetes |
Data from Schunkert et al.34
An unforeseen discovery: the chromosome 9p21 locus and CHD risk
The region at chr9p21 was an unexpected site for a CHD risk locus, and has prompted much research into the biological mechanisms underlying the finding. Furthermore, the chr9p21 locus appears to be GWAS ‘hotspot’, showing an association with other cardiovascular disease (CVD) phenotypes such as type 2 diabetes,35 abdominal aortic aneurysm (AAA) and intracranial aneurysm.36 This non-specific effect does not appear to be mediated by effects of the variants on intermediate phenotypes such as circulating lipid concentrations, blood pressure, carotid intima media thickness or biomarkers of inflammation, suggesting an alternative mechanism of action.37,38 The variants lie distant from known genes: the nearest protein coding genes are CDKN2A (150 kb) and CDKN2B (118 kb), which encode inhibitors of cellular senescence involved in controlling cellular proliferation and apoptosis. A link has, however, been identified between the risk variants and the recently annotated non-coding RNA (ncRNA), ANRIL (antisense ncRNA in the INK4 locus).39,40 ncRNAs can alter the expression of protein-coding genes through gene silencing, DNA methylation, chromatin remodelling and RNA interference.41 Early functional studies have shown that the chr9p21 locus has an enhancer activity in the primary human aortic smooth muscle cells and that pathways involved in cellular proliferation were upregulated in risk allele carriers,42 while the targeted deletion of this region in a mouse model leads to increased expression of CDKN2A and CDKN2B and aortic smooth muscle cells that displayed a highly proliferative phenotype.43 The chr9p21 region has also been found to contain over 30 predicted enhancer sites that play a regulatory role in the expression of both nearby and distant gene sets.44 Specifically, variants associated with CHD alter binding sites for the transcription factor STAT1, which regulates the expression of CDKN2A and CDKN2B, MTAP (methylthioadenosine phosphorylase, an enzyme with an important role in polyamine metabolism) and IFNA21 (interferon alpha-21). Although the mechanisms by which these variants exert their effect on CHD risk are becoming clearer, the translational impact of these discoveries in respect of identifying novel treatment targets is likely to lie some way into the future.
Implications of GWAS findings for prediction of CHD risk
The advances in our understanding of the genetic susceptibility to CHD have naturally led to the question of whether this information can be used to predict an individual's risk of disease. Prediction of future disease risk is an important component of the clinical management of CHD chiefly because it allows the identification of individuals at high risk in order to intervene to change modifiable risk factors (e.g. smoking status, blood pressure, exercise) and reduce their subsequent risk of disease.
Of the many SNPs identified to date that are associated with CHD, most have modest effects on disease risk, and so have small incremental effects when included in a risk score incorporating a large number of variants. Many have questioned whether the use of these SNPs has a clinical value in disease prediction. Several metrics for the success of a predictive instrument exist, each with its advantages and limitations. Perhaps the most widely used is the area under the receiver-operating characteristic curve (ROC), quantified by the C-statistic, where a value of 0.5 indicates no discrimination between those that develop a CHD event and those that remain free from disease, and 1.0, perfect discrimination. In a recent study, the addition of 101 SNPs associated with CHD risk to a traditional risk prediction score (comprising non-genetic markers including age, blood pressure and smoking status) yielded no change in C-statistic.45 Thus, the clinical utility of genetic information in CHD prediction appears questionable. However, it is possible that as our understanding of the genetic architecture of CHD grows, and as more genes that independently associate with disease are identified, genetic data may find a role in prediction in the clinical setting. In particular, the potential of combining information from many SNPs in a gene score appears promising. Before this time, we believe that the direct-to-consumer provision of genetic tests should be regulated to protect consumers.46
Exploitation of GWAS findings for prevention, therapeutics and pathophysiology
While the variants that have been discovered to date may not have immediate clinical utility in predicting the occurrence of CHD, this does not preclude other important translational applications. Indeed, while it is true that the variants identified for their associations with risk of CHD do not explain disease risk entirely, they do have substantial potential to reveal novel pathogenic mechanisms, and to suggest targets for prevention and therapy.47
The principle of Mendelian randomization (MR) underlies much research of this type and exploits some of the fundamental properties of genetic variation to identify causal factors in complex disease.48,49 In epidemiological terms, genotype is a unique population ‘exposure’ for three reasons: first, according to Mendel's second law,50 genotype at a given locus is allocated at random during the random segregation of alleles at gametogenesis; secondly, the central dogma of molecular biology51 dictates that information flows in only one direction from a DNA sequence through messenger RNA (mRNA) and protein to a more complex phenotype (e.g. CHD); and, finally, unlike many exposures associated with CHD risk such as diet and smoking behaviour, an individual's exposure to their genotype is constant, lifelong and unchanged by the presence of disease. These three features help MR to overcome two important limitations that beset traditional observational epidemiology—confounding and reverse causation. In the same way as random allocation to the treatment or control arm avoids confounding in a randomized controlled trial (RCT), so too does the random allocation of genotype in an MR study.52 An apparent risk factor associated in an observational study with disease risk may, in fact, be a consequence of the disease process and that association represents reverse causation. The unidirectional flow of information from genotype to phenotype avoids this (Fig. 2).
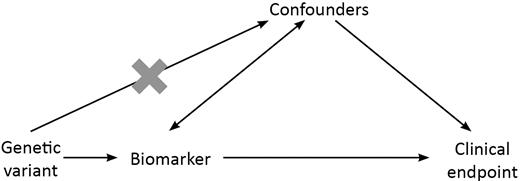
The MR paradigm. The effect on disease risk of a genetic instrument associated with the biomarker is evaluated whilst avoiding confounders of the observed relationship between the biomarker and disease. The paradigm rests on three assumptions: (i) the genetic variant is associated with the biomarker; (ii) the genetic variant is independent of confounders of the biomarker-disease relationship and (iii) the genetic variant exerts its effect on disease risk only through the action of the biomarker.
The application of MR for evaluating the causal relevance of many of the risk factors associated with CVD risk, of which many have a still uncertain role, is illustrated in Figure 2. Using genetic variants (or better a combination of variants in a ‘gene score’) identified for their associations with a risk factor (e.g. a biomarker of inflammation such as C-reactive protein, CRP), the role of that risk factor—causal or not—in the development of the disease may be determined. For example, a causal role for CRP in CHD pathogenesis has long been proposed based on observational associations,53 but in the absence of a specific CRP-lowering drug licensed for use in humans, that role could neither be confirmed nor refuted using an RCT. A number of studies have identified common variants in the gene encoding CRP (CRP, chr1q23.2) that are associated with circulating CRP concentration.54–57 These variants have been used as unconfounded, unbiased ‘instruments’ for CRP concentration in large population-based MR studies.57–60 In these studies, the association between the carriage of a CRP-raising allele and risk of CHD events is estimated. Since the genetic variants are associated causally with CRP concentrations, and are free from confounding and reverse causation, an association of the variants with CHD risk would support a causal role for CRP in the development of CHD events (Fig. 2). In contrast, all published MR studies examining the role of CRP in CHD pathogenesis, with samples of up to 194 000 individuals, have found null associations of genetic variants in CRP with risk of disease, leading to the conclusion that the role of CRP is very unlikely to be causal.57–60
In addition to its utility for investigating endogenous biomarkers, MR has been used to address the causal relevance of environmental and behavioural risk factors, and examples of MR studies of relevance to CVD are shown in Table 3. There are, of course, limitations to the approach, and it is important that the SNP or gene score used as a genetic instrument is associated only with the trait being examined, and does not also influence the trait or disease process through additional, potentially confounding pathways. For example, the APOA5 SNP used to confirm the causal relationship between higher triglyceride levels and CHD risk is also weakly associated with lower levels of HDL-C, although in this case the large effect of this SNP on CHD is unlikely to be explained by its small effect on HDL-C.61
MR studies where causality for a biomarker in CHD has been confirmed or refuted.
| Risk factor | Genetic instrument locus | Finding(s) |
|---|---|---|
| Confirmed | ||
| Plasma glucose | Score (GCKR, G6PC2, GCK, SLC30A8, MTNR1B) | Strong association of gene score with carotid artery wall thickness suggests a causal role for glucose in atherosclerosis73 |
| Triglyceride-mediated pathways | APOA5 | Although not exclusive in its association with triglycerides, an APOA5 variant was strongly associated with that lipid fraction and CHD risk, indicating that pathways involving triglycerides are causal factors61 |
| LDL-C | LDLR | As expected, a variant in LDLR associated with plasma LDL-C concentration is also associated with CHD risk, recapitulating the established causal role of LDL-C in CHD74 |
| Lipoprotein(a) | LPA | Concordant associations of LPA variant with lipoprotein(a) (Lp(a)) levels and CHD risk suggest a causal role for Lp(a) in CHD75 |
| Refuted | ||
| CRP | CRP | A number of large studies have shown CRP-associated variants in CRP have no association with CHD risk suggesting CRP is very unlikely to play a causal part in CHD57–60 |
| Fibrinogen | FGB | Variants in FGB associated with fibrinogen levels show no association with CHD risk and therefore fibrinogen is unlikely to determine CHD risk76 |
| Risk factor | Genetic instrument locus | Finding(s) |
|---|---|---|
| Confirmed | ||
| Plasma glucose | Score (GCKR, G6PC2, GCK, SLC30A8, MTNR1B) | Strong association of gene score with carotid artery wall thickness suggests a causal role for glucose in atherosclerosis73 |
| Triglyceride-mediated pathways | APOA5 | Although not exclusive in its association with triglycerides, an APOA5 variant was strongly associated with that lipid fraction and CHD risk, indicating that pathways involving triglycerides are causal factors61 |
| LDL-C | LDLR | As expected, a variant in LDLR associated with plasma LDL-C concentration is also associated with CHD risk, recapitulating the established causal role of LDL-C in CHD74 |
| Lipoprotein(a) | LPA | Concordant associations of LPA variant with lipoprotein(a) (Lp(a)) levels and CHD risk suggest a causal role for Lp(a) in CHD75 |
| Refuted | ||
| CRP | CRP | A number of large studies have shown CRP-associated variants in CRP have no association with CHD risk suggesting CRP is very unlikely to play a causal part in CHD57–60 |
| Fibrinogen | FGB | Variants in FGB associated with fibrinogen levels show no association with CHD risk and therefore fibrinogen is unlikely to determine CHD risk76 |
MR studies where causality for a biomarker in CHD has been confirmed or refuted.
| Risk factor | Genetic instrument locus | Finding(s) |
|---|---|---|
| Confirmed | ||
| Plasma glucose | Score (GCKR, G6PC2, GCK, SLC30A8, MTNR1B) | Strong association of gene score with carotid artery wall thickness suggests a causal role for glucose in atherosclerosis73 |
| Triglyceride-mediated pathways | APOA5 | Although not exclusive in its association with triglycerides, an APOA5 variant was strongly associated with that lipid fraction and CHD risk, indicating that pathways involving triglycerides are causal factors61 |
| LDL-C | LDLR | As expected, a variant in LDLR associated with plasma LDL-C concentration is also associated with CHD risk, recapitulating the established causal role of LDL-C in CHD74 |
| Lipoprotein(a) | LPA | Concordant associations of LPA variant with lipoprotein(a) (Lp(a)) levels and CHD risk suggest a causal role for Lp(a) in CHD75 |
| Refuted | ||
| CRP | CRP | A number of large studies have shown CRP-associated variants in CRP have no association with CHD risk suggesting CRP is very unlikely to play a causal part in CHD57–60 |
| Fibrinogen | FGB | Variants in FGB associated with fibrinogen levels show no association with CHD risk and therefore fibrinogen is unlikely to determine CHD risk76 |
| Risk factor | Genetic instrument locus | Finding(s) |
|---|---|---|
| Confirmed | ||
| Plasma glucose | Score (GCKR, G6PC2, GCK, SLC30A8, MTNR1B) | Strong association of gene score with carotid artery wall thickness suggests a causal role for glucose in atherosclerosis73 |
| Triglyceride-mediated pathways | APOA5 | Although not exclusive in its association with triglycerides, an APOA5 variant was strongly associated with that lipid fraction and CHD risk, indicating that pathways involving triglycerides are causal factors61 |
| LDL-C | LDLR | As expected, a variant in LDLR associated with plasma LDL-C concentration is also associated with CHD risk, recapitulating the established causal role of LDL-C in CHD74 |
| Lipoprotein(a) | LPA | Concordant associations of LPA variant with lipoprotein(a) (Lp(a)) levels and CHD risk suggest a causal role for Lp(a) in CHD75 |
| Refuted | ||
| CRP | CRP | A number of large studies have shown CRP-associated variants in CRP have no association with CHD risk suggesting CRP is very unlikely to play a causal part in CHD57–60 |
| Fibrinogen | FGB | Variants in FGB associated with fibrinogen levels show no association with CHD risk and therefore fibrinogen is unlikely to determine CHD risk76 |
Predicting drug response in individuals: pharmacogenetics
As early as the 1950s, differences in drug response were recognized to arise from genetic variation—so-called ‘pharmacogenetics’. It has been suggested that understanding pharmacogenetics would herald an era of personalized treatment plans, whereby an individual's genotype could be used to predict response to a drug and/or adverse drug reactions (ADRs). More recently, it has become clearer that, in general, differences in response to a drug are of little clinical importance, since all patients have some response, for example to a lipid-lowering or blood pressure-lowering agent, and dose can be easily titrated to achieve the desired effect.
There are two principal means by which genetic polymorphisms can influence responses to drug treatment: first, variation in the drug target, and second, variation in drug metabolism pathways. In the former case, variants in the gene encoding the protein target of a drug may alter drug binding, resulting in reduced drug efficacy. In the context of drugs for CHD prevention, a treated patient who carries a variant that reduces drug efficacy may be at higher risk of CHD compared with another who does not. The latter scenario (variation in drug metabolism pathways) frequently concerns variants in genes encoding cytochrome P450 (CYP) or UDP-glucuronosyltransferase enzymes responsible for drug metabolism (i.e. a pharmacokinetic property), although variation in genes coding for proteins involved in intestinal absorption and blood transport of compounds may also be relevant. Genetic variation might thus alter the exposure to the active drug, which could influence both the intended and adverse effects of the agent. If the drug has mechanism-based adverse effects, such as the anticoagulant warfarin (excess circulating levels of which can cause life-threatening haemorrhage), information about gene variants that alter drug metabolism could be used to identify patients at higher risk of an unwanted effect. For example, variants in CYP2C9 and/or the vitamin K epoxide reductase gene, VKORC1, have been used to predict ‘slow metabolizers’ of warfarin, such that tailored dosing guidelines could be implemented.62 Despite the theoretical benefits of such an approach, there is little evidence to support either efficacy or improved cost-effectiveness. With the development of newer drugs63 that do not have such a narrow therapeutic index, it is possible that drug development may overtake the clinical utility of this pharmacogenetic test.64
Another related issue is that many of the initial positive reports in the pharmacogenetic literature may have been the result of false-positive findings from small studies. For example, it has been reported that carriers of a common variant in the KIF6 gene have less cardiovascular risk reduction than non-carriers when treated with a statin (reviewed in ref. 65), and on this basis, KIF6 testing has been proposed as a clinically useful pharmacogenetic test. However, analysis in the Heart Protection Study, with over 18 000 participants randomized to placebo or simvastatin 40 mg, demonstrated conclusively a similar reduction in LDL-C and a similar reduction in CHD risk in subjects irrespective of KIF6 genotype.66
A more important clinical application of pharmacogenetics would be to prevent ADRs, some of which can be life threatening, as well as costly to health services. Currently, there are only a few examples of useful pharmacogenetic tests to avoid ADRs, and although not in the field of CHD, the routine clinical use in the UK of HLA-B*5701 genotype to predict hypersensitivity reactions to HIV medications is an important example.67 In CVD, a serious but rare ADR in people treated with statins is a life-threatening form of myopathy, occurring in roughly 1/10 000 patients. Recently, a GWA study identified a variant in the SCLO1B1 gene strongly associated with this risk; individuals homozygous for the variant had a 17-fold higher risk than non-carriers.68 However, at present there is not a policy to use this genetic test before the commencement of statin therapy. Finally, a therapeutic area in which pharmacogenetics may hold promise is in targeting the use of biological agents (e.g. monoclonal antibodies) that have potentially serious adverse effects and high costs that prevent universal treatment in resource-limited healthcare systems such as the UK National Health Service.
While in the case of hypersensitivity reactions (for which identified gene variants may have large phenotypic effects, expediting clinical translation), and in the field of cancer medicine, pharmacogenetics has found a place in clinical practice, currently, the routine use of pharmacogenetics to inform clinical prescribing in cardiovascular disease is of unproven clinical or cost benefit. Whether using genotype to predict drug response will ever yield better results than a universal treatment strategy is debatable, but improvements are needed in the conduct of pharmacogenetic studies to reduce bias and maximize reliability in the resulting data.69,70
Future prospects: next generation sequencing technology
With the development of ‘next generation’ genomic sequencing (NGS) technologies, it is now possible to obtain at relatively high speed and low cost, whole genome sequence data for an individual, with both cost and time requirements declining rapidly. NGS technology promises to answer many of persisting questions in the genetics of CHD, for example, identification of the causal variants/mutations at GWAS-detected loci, and determining the role of rare variants in the genetics of common complex diseases. The NGS method uses high-density oligonucleotide microarrays to capture short genome segments by hybridization. These regions can include the whole genome, all the exons of a gene or series of genes (for example, all the FH-causing genes), or even longer, complete regions,71 which are then sequenced.
At first glance, this information appears likely to be valuable for predicting individual patient risk, but there are several limitations that warrant consideration. The first is the reliability of assigning true causality for the trait or disease to the detected genetic sequence change, an issue encountered daily in clinical genetic diagnostic laboratory. Exome sequencing in healthy individuals has revealed that ‘loss of function’ variants are a surprisingly common finding, for example, those that cause a truncation in encoded proteins, with, on average, every sequenced individual carrying more than 100 such variants.72 Computer algorithms are available to assist with assigning impact of identified variants based on structure–function predictions, but definitive demonstration of causality may require expensive and time-consuming in vitro expression assays. The second challenge is the quantification of the degree of CHD risk that can be assigned to the identified variant. Meta-analyses have reported odds ratios in the range of 1.2–1.4 associated with common functional variants, but robust proof of these effects has required pooling of many studies, and the combined effects of carrying more than one such variant have not been explored. Whilst a number of statistical methods have been described to deal with these challenges, successes in large-scale re-sequencing projects have so far been largely limited to rare, monogenic diseases. It is likely that many of the challenges encountered in GWAS will be experienced, perhaps to a greater degree, in NGS studies, particularly those surrounding statistical power. The availability in the next few years of large data sets from high-resolution sequencing projects, for example from the 1000 Genomes Project (www.1000genomes.org), may, however, help to overcome some of these challenges.
Conclusions
Genomic advances in the past 10 years have catalysed growth in our understanding of the genetic basis of CHD. We are, however in the very early phases of the clinical translation. Techniques such as MR provide unprecedented potential to understand disease causality and identify drug targets, and are already bearing fruit. However, other areas such as the useful incorporation of genetic information into predicting an individual's risk of CHD or the routine clinical use of pharmacogenetics in stratifying medical therapy will take longer to translate, although the use of gene scores appears promising. While we equip ourselves with the statistical and technological advances to make this possible, we should not lose focus of the dramatic leaps the scientific community has made in uncovering new information which will be of undoubted benefit for preventing disease in future generations.
Funding
MVH is funded by a Population Health Scientist Fellowship from the Medical Research Council (G0802432). The British Heart Foundation fund SCH as a Clinical Training Fellow (FS/11/16/28696) and SEH (RG08/008). DIS is supported by a UK Medical Research Council Doctoral Training Award. This work was undertaken at UCLH/UCL who received a proportion of funding from the Department of Health's NIHR Biomedical Research Centres funding scheme.
Relationship with Industry
SEH is the Medical Director of the UCL genetic testing company Storegene and has received honoraria for speaking at educational meetings with a pharmaceutical sponsor, but has donated these in whole or part to various medical charities.

{kind=link}
{kind=link}